En este artículo analizamos el significado de entropía y su importancia en termodinámica, tanto en el universo como dentro de un sistema.
La entropía es una medida de todas las configuraciones posibles (o microestados) de un sistema. La entropía suele describirse como el grado de desorden de un sistema. Los sistemas ordenados tienen menos configuraciones disponibles, por lo que su entropía es menor. Es importante destacar que la entropía es una función de estado, como la temperatura o la presión, y no una función de trayectoria, como el calor o el trabajo. Esto significa que, cuando un sistema cambia de entropía, el cambio sólo depende de las entropías de los estados inicial y final, y no de la secuencia («trayectoria») seguida entre los estados.
La letra «S» se utiliza para representar la entropía.
Como veremos más adelante, la entropía es muy útil para químicos y físicos a la hora de determinar la espontaneidad de un proceso.
Alta vs. baja entropía
Un sistema con baja entropía implica partículas ordenadas y movimiento dirigido. Pensemos en una casa. La masa que compone la casa está ordenada y es exacta para formar las paredes y los muebles. Cualquier energía mecánica de movimiento, como el agua y el gas moviéndose a través de las tuberías, permanece ordenada y dirigida. La energía calorífica también permanece controlada, con ciertas bolsas frías, como el frigorífico, y calientes, como el horno, con temperaturas diferentes que no se propagan al resto de la casa.

En química, una masa sólida de cristal constituye otro buen ejemplo de sistema entrópicamente bajo. La energía reticular del cristal limita el movimiento de sus partículas, lo que da como resultado una forma perfectamente geométrica.
Por el contrario, un sistema con una entropía alta implica una masa y una energía muy dispersas. Pensemos en un bosque. La masa de los árboles, plantas, rocas y animales permanece aleatoria y muy dispersa. Del mismo modo, el movimiento y el calor se dispersan, dando lugar a una temperatura relativamente constante y a movimientos impredecibles de árboles y animales.

En química, un gas constituye otro buen ejemplo de sistema entrópico. La atracción relativamente baja entre las partículas del gas permite que cada molécula se mueva libremente, lo que da lugar a una dispersión aleatoria.
Definición estadística
La principal forma de cuantificar el orden de la materia y la energía consiste en sumar los microestados que puede tener un sistema determinado. Los químicos definen un microestado como una disposición específica de la materia y la energía. Naturalmente, los sistemas ordenados, entrópicamente inferiores, tienen menos microestados posibles que los sistemas desordenados, entrópicamente superiores.
Este enfoque estadístico implica la siguiente fórmula logarítmica natural que relaciona la entropía con los microestados:
S = klnW
S = Entropía (J/K)
k = Constante de Boltzman (1.381*10-23 J/K)
W = Número de Microstates Posibles
Obsérvese que la entropía se expresa en julios por Kelvin.
Sin embargo, contar los microestados individuales sigue siendo imposible en la mayoría de los sistemas. Por tanto, esta definición es la más útil para calcular los microestados de un sistema a partir de valores entrópicos conocidos. En estos casos, los químicos suelen calcular la entropía utilizando la definición termodinámica.
Definición termodinámica
En lugar de tratar con microestados, la mayoría de los químicos miden los valores entrópicos utilizando la calorimetría. Así, los químicos pueden definir la entropía termodinámicamente, utilizando el flujo de calor y la temperatura del sistema:
dS = dqrev / T
dS = Cambio pequeño en entropía (J/K)
dqrev = Cambio pequeño en calor (J)
T = Temperatura (K)
Esta fórmula de la entropía suele ser la más útil para medir el cambio entre dos estados:
ΔS = – ∫if dqrev / T
Es importante señalar que el calor utilizado para calcular la entropía es el que se desprende o absorbe si el cambio en cuestión se ha realizado de forma reversible. Aunque el calor suele ser una función de trayectoria, sólo existe un camino reversible entre dos estados, lo que la convierte en una función cuasiestatal, como la entropía. Es importante destacar que seguimos utilizando el calor reversible incluso cuando calculamos el cambio de un cambio irreversible entre dos estados.
Podemos simplificar aún más la ecuación anterior dependiendo de si la temperatura cambia entre los dos estados:
ΔS = qrev / T ΔS = qrev (1/Ti – 1/Tf)
(Izquierda: Cambio de entropía si la temperatura permanece constante. Derecha: Cambio de entropía si cambia la temperatura).
La entropía es importante para los químicos y los físicos porque determina la espontaneidad de los procesos. Para entender mejor lo que esto significa, tenemos que fijarnos en la Segunda Ley de la Termodinámica.
La Segunda Ley establece que la entropía del universo siempre aumenta. Cualquier cambio físico debe aumentar o no el desorden global del universo. No es posible ningún proceso que tenga el efecto global de ordenar y dirigir la masa y la energía del universo.
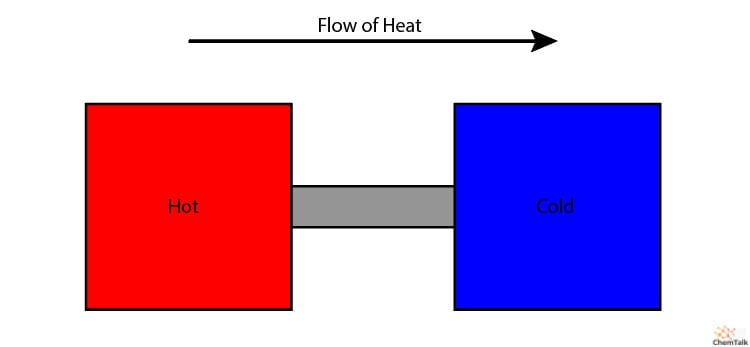
Teniendo esto en cuenta, digamos que tenemos dos trozos de metal, uno caliente y otro frío. Ahora, coloquemos un puente metálico conductor, permitiendo que el calor fluya entre los dos. El calor fluirá entre el metal caliente y el frío de forma espontánea, es decir, sin intervención externa.
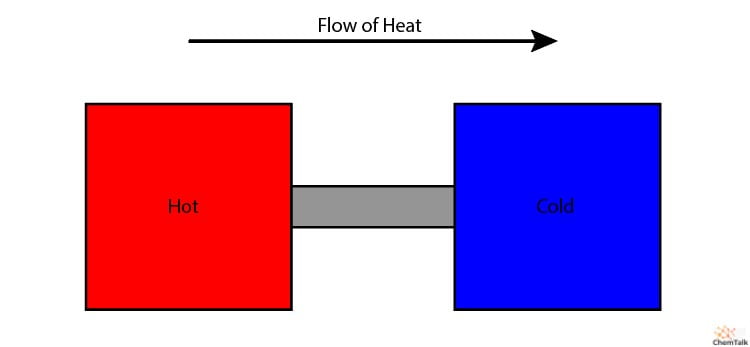
¿Por qué no fluiría el calor del metal frío al caliente, aumentando aún más su temperatura? Al fin y al cabo, este movimiento seguiría cumpliendo la Primera Ley, ya que no se crea ni se destruye energía.
La Segunda Ley explica por qué esto no ocurre. El nivel entrópico del sistema disminuiría al concentrarse el calor en el metal caliente, pero aumentaría si la energía térmica se distribuye uniformemente por ambos metales. Así, el calor sólo fluye del metal caliente al frío, para permitir que la entropía del universo siga aumentando.
Es importante señalar que, mientras la entropía del universo debe aumentar sin excepción, la del sistema puede disminuir espontáneamente.
Por ejemplo, tomemos un motor térmico. En general, un motor térmico funciona tomando energía calorífica y convirtiéndola en trabajo. Esta conversión implica una disminución entrópica del sistema, ya que la energía desordenada del calor se convierte en movimiento ordenado. A pesar de ello, los motores térmicos siguen funcionando espontáneamente en el mundo real. La razón es que cierta cantidad de calor del motor se libera al universo (es decir, a un sumidero frío). Este calor, como resultado de la distribución en el universo, implica un aumento de la entropía que contrarresta la disminución procedente del trabajo del motor.
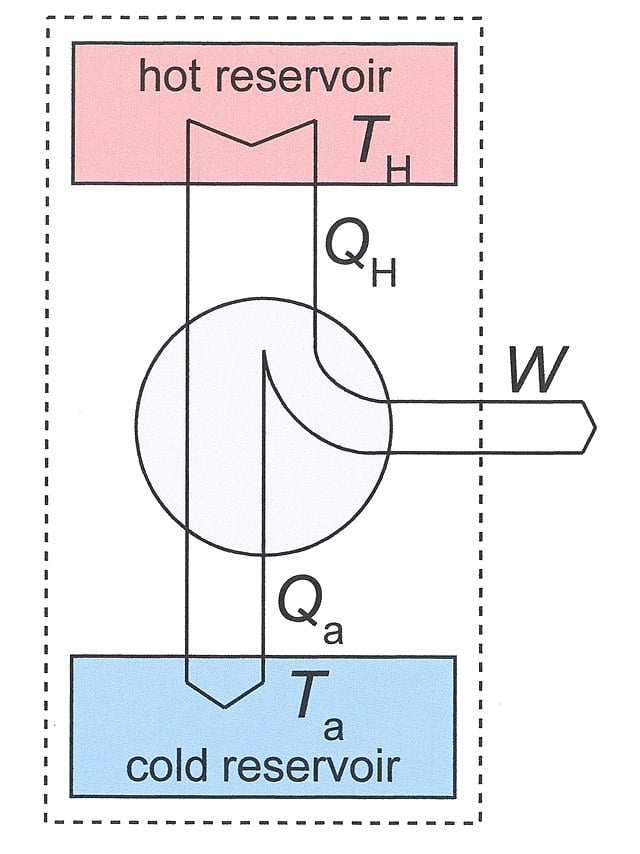
No toda la energía térmica del depósito caliente (QH) se transforma en trabajo (W). Una parte se libera en forma de calor (Qa). Fuente.
Por mandato de la Segunda Ley, este calor debe ser liberado por el motor para evitar la disminución de la entropía universal.
Volviendo a nuestros primeros ejemplos, la Segunda Ley explica por qué los bosques no se convierten espontáneamente en casas, sino que, dado el tiempo suficiente, las casas se degradan espontáneamente hasta convertirse en bosques. Sin embargo, los bosques siguen convirtiéndose en casas en el mundo real. Como sabemos ahora, esto se debe a que la energía química se gasta, liberando calor en el universo, de los cuerpos humanos que realizan el trabajo necesario.
Entropía ante entalpía
En nuestros ejemplos anteriores, los químicos utilizan el término «entalpía» para describir este calor desprendido por un proceso que disminuye la entropía. La entalpía es un concepto termodinámico importante, pero distinto, para determinar la espontaneidad termodinámica. Si quieres saber más sobre la entalpía, consulta este artículo.
Energía libre de Gibbs
Así que hemos cubierto que los cambios entálpicos negativos pueden ocurrir, pero solo si el calor, en forma de entalpía, se libera para elevar la S universal. Para comprender matemáticamente la relación entre la espontaneidad y estas dos variables, debemos entender una tercera: la energía libre de Gibbs (G). La siguiente ecuación ilustra la relación entre entropía, entalpía (H), energía libre de Gibbs y temperatura:
ΔG = ΔH – TΔS
ΔG = Cambio en Energía Libre de Gibbs (kJ/mol)
ΔH = Cambio en Entalpía (kJ/mol)
ΔS = Cambio en Entropía (kJ/mol)
T = Temperatura
Si en una reacción química interviene ΔG < 0, la reacción es espontánea. Por tanto, si ΔS< 0, la reacción sólo puede producirse espontáneamente si ΔH < 0, lo que implica calor desprendido por la reacción. Para saber más sobre la energía libre de Gibbs, consulta este artículo.
Problema 1
El Sistema 1 tiene un valor de entropía 0.00000000000000000000001J/K (1 * 1022J/K) mayor que el Sistema 2. ¿Cuál es la relación de microestados entre el Sistema 1 y el Sistema 2?
Problema 2
Un sistema a 50℃ libera 23kJ calor en a través de un proceso reversible, bajando su temperatura a 32℃. ¿Cuál es el cambio de entropía?
1: 1396 : 1
2: 1971/K
The post ¿Qué es la entropía? appeared first on ChemTalk.

En este tutorial aprenderás la definición y la ecuación de la entalpía. Además, aprenderás sobre algunas de sus aplicaciones, ya que se relaciona con la termodinámica.
La entalpía (H) tiene que ver con la termodinámica; es una función de estado utilizada en sistemas químicos y biológicos. Esto significa que la entalpía sólo depende de la energía final, la presión y el volumen, y no de la trayectoria que siguió el sistema para llegar al estado final. En la mayoría de los sistemas químicos basados en soluciones, donde la presión y el volumen permanecen constantes, la entalpía es igual a la energía interna del sistema, lo que significa que el cambio en la entalpía es el calor absorbido o liberado en un proceso termodinámico, como una reacción química. Esto se debe a la ley de conservación de la energía. Si la presión o el volumen cambian, por ejemplo si se produce un gas durante una reacción, entonces el cambio de entalpía ya no será igual al cambio de calor.
Esta propiedad es la suma de la energía interna de un sistema y el producto de la presión y el volumen.
H = U + pV
H : Enthalpy
U : Internal energy of the system
p: Pressure
V: Volume
You also may see pV expressed as W, known as work.
H = U + W
La unidad de medida SI para la entalpía es el julio (J). Sin embargo, a veces puedes ver las unidades de caloría o unidad térmica británica (BTU).
Un factor que entra en juego a la hora de determinar la entalpía, aunque no se vea en las ecuaciones anteriores, es la temperatura. Los mismos reactantes en una reacción pueden variar en la cantidad de calor que pueden transferir cuando están a diferentes temperaturas. Además, los distintos reactantes pueden estar en distintas fases de la materia a distintas temperaturas ambiente, lo que también puede influir en la transferencia de calor.
Cuando un proceso es endotérmico, absorbe calor al sistema. Esto hace que el valor de ΔH sea positivo. Por el contrario, cuando un proceso es exotérmico, libera calor al entorno. Recuerde que, aunque parezca que hay un cambio de energía, estas reacciones, como todas las reacciones químicas, obedecen a la ley de conservación de la energía.
En el mundo real se producen reacciones químicas en todas partes y hay varios ejemplos de entalpía en la vida cotidiana. Un ejemplo son los paquetes calientamanos portátiles. Estos paquetitos sufren una reacción de oxidación del hierro a presión constante en un sistema cerrado, razón por la cual, cuando se rompe el paquetito, se calienta rápida y fácilmente. Este es un ejemplo de reacción exotérmica porque el sistema libera energía en forma de calor. Otro ejemplo de entalpía puede verse en los compresores de los refrigeradores. Aquí se produce una reacción de vaporización, también bajo la condición exacta de presión constante. Se está utilizando y absorbiendo energía en el sistema en forma de calor a medida que los productos químicos refrigerantes se vaporizan en una cuestión endotérmica.
Problema 1
Cuando el vapor de agua se condensa en líquido, se libera calor al ambiente. Por tanto, ¿la condensación es exotérmica o endotérmica?
Problema 2
Considera la ecuación para el cambio en la energía libre de Gibbs:
ΔG = ΔH – TΔS
Si una determinada reacción química implica un cambio negativo en la entropía (ΔS < 0) y un cambio negativo en la energía libre de Gibbs (ΔG < 0), ¿es exotérmica o endotérmica? (Pista: como la temperatura se expresa en grados Kelvin, sabemos T < 0)
1: Exotérmica
2: Exotérmica
Para repasar algunos problemas de ejemplo detallados sobre la entalpía, consulta nuestro artículo sobre la Ley de Hess.
Para aprender a calcular la entalpía, consulta varias técnicas diferentes en la página de Calcular la Entalpía.
The post ¿Qué es la entalpía? appeared first on ChemTalk.

En este artículo, distinguimos entre variables y funciones de Estado vs. Trayectoria, describiendo cada tipo y explicando cómo convertir entre ellas.
En química, nos gusta utilizar distintas variables y propiedades para describir sistemas termodinámicos. Cuando conocemos cantidades como la presión, la temperatura, la entalpía y la energía libre de Gibbs, podemos predecir con exactitud el comportamiento físico y químico. Los químicos físicos dividen estas variables en dos categorías en función de sus propiedades matemáticas y conceptuales. Estas dos categorías son las variables de estado y las variables de trayectoria. Para comprender gran parte de la termodinámica, es importante comparar y contrastar las variables de estado con las variables de trayectoria.
Las variables de estado son magnitudes que dependen del estado actual de un sistema. Los estados anteriores o los cambios en el sistema no afectan a las variables de estado. Muchos de los parámetros físicos y químicos más básicos de un sistema son variables de estado. Algunos ejemplos son la temperatura, la presión, el volumen, los moles y la concentración. Muchos parámetros termodinámicos avanzados, como la entropía, la energía libre de Gibbs, la energía cinética y la energía interna, también son variables de estado.
Cuando hablamos de sustancias concretas, las variables de estado pueden ser extensivas o intensivas. Las variables de estado extensivas dependen de la cantidad de esa sustancia, como la masa o el volumen. Las variables de estado intensivas, por el contrario, no cambian en función de la cantidad de la sustancia, como la densidad o la electronegatividad. El tungsteno, por ejemplo, puede ocupar distintas masas o volúmenes, pero su densidad y electronegatividad permanecen inalterables.
Funciones de estado
Para ilustrar cómo cambian las variables de estado (extrínsecas), los químicos físicos utilizan funciones de estado. Estas funciones sólo dependen de las variables de estado y de las constantes termodinámicas. Utilizando la Ley de los Gases Ideales, podemos resolver para la presión, generando la siguiente función de estado para la presión:
P = nRT / V
Dado que las variables de estado sólo dependen del estado actual, sólo necesitamos los estados inicial y final para calcular el cambio en una función de estado. Los estados intermedios, o «trayectos», entre los dos estados no entran en el cálculo.
ΔP = Pfinal -Pinitial
De este modo, las variables de estado como la entropía, la entalpía y la energía libre de Gibbs pueden calcularse mediante la Ley de Hess, que genera funciones de estado restando la suma de los valores del producto menos los de los reactantes. Estas funciones producen el cambio en esa variable termodinámica para la reacción.
ΔHrxn = ∑ΔHf,productos – ∑ΔHf,reactantes
ΔSrxn = ∑Sproductos – ∑Sreactantes
ΔGrxn = ∑ΔGf,productos – Δ∑Gf,reactantes
Lista de variables de estado importantes
T – Temperatura
mol – Moles
m – Masa
p – Densidad
V – Energía potencial
S – Entropía
H – Entalpía
P – Presión
V – Volumen
x – Fracción molar
MM – Peso molecular
K – Energía cinética
G – Energía libre de Gibbs
U – Energía interna
En cambio, las variables de trayectoria dependen de la secuencia concreta que convierte un estado en otro. Las variables de trayectoria no describen un estado termodinámico concreto, sino un proceso, como una reacción química. En termodinámica, el calor y el trabajo son las variables de trayectoria más importantes.
Funciones de trayectoria
El contexto clásico en el que los químicos utilizan las variables de trayectoria es el de la expansión y compresión isotérmica de gases. Específicamente, cuando se tiene un pistón lleno de gas, se puede comprimir el gas, aumentando así su presión. Esto se debe a que la compresión reduce el volumen mientras que la temperatura permanece igual (como implica el término «isotérmico»). Lo sabemos por nuestra función de estado para la presión.
P = nRT / V
Este acto de compresión cuenta como la realización de un trabajo sobre el gas. Puedes calcular el trabajo según la siguiente función:
w = – ∫ViVf PdV
El trabajo: Un paso frente a dos pasos
Nuestra expresión para el trabajo depende del volumen inicial y final del gas, así como de la presión. Si realizas la compresión en un solo paso, consideramos que la presión del gas no cambia. Los químicos llaman a esto compresión «irreversible». Con presión constante, puedes integrar para generar la siguiente fórmula:
w = –P(Vf – Vi)
Sin embargo, si la compresión se produce en varios pasos separados, acabamos con una cantidad de trabajo diferente. Específicamente, digamos que se comprime en dos pasos, alcanzando algún volumen intermedio (Vint) antes de alcanzar el volumen final. La presión del gas alcanza cierto equilibrio en ese volumen intermedio (Pint), lo que afecta a la segunda compresión. Ambos pasos siguen siendo irreversibles, ya que ocurren (básicamente) al instante.
w = -Pi(Vint – Vi) – Pint(Vf – Vint)
Dado que el Pint es mayor que Pi, después de una compresión parcial, cabría esperar que se requiría menos trabajo para comprimir en dos pasos que en uno. Permitir que el gas alcance el equilibrio antes de una nueva compresión alivia gran parte de la tensión del gas. Si se realiza una compresión en pasos infinitamente pequeños, se alcanza el trabajo mínimo necesario para comprimir el gas.
Todo esto para subrayar que, aunque el gas comprimido acabe en el mismo estado final, el trabajo necesario para realizar la compresión cambia en función del camino concreto que se tome.
Diagrama de un motor térmico en el que intervienen dos variables de recorrido (trabajo y calor) y una variable de estado (temperatura).
En termodinámica, las funciones de estado a menudo implican variables de trayectoria, y las funciones de trayectoria a menudo implican variables de estado. Esto puede no tener mucho sentido al principio, ya que los cambios de estado no deberían depender de los detalles de la trayectoria. En la mayoría de los casos, sin embargo, estas relaciones simplemente demuestran que un número limitado de caminos son posibles para un cambio de estado dado.
Por ejemplo, veamos el cambio en la energía interna, una variable de estado. Debido a la Primera Ley de la Termodinámica, sabemos que el cambio en la energía interna depende del calor y del trabajo.
ΔU = w + q
En esta fórmula, podemos presenciar que nuestras variables de trayectoria de trabajo y calor deben entonces sumar el cambio en la energía interna. Esto pone una limitación termodinámica a nuestras posibles trayectorias, dado un cierto cambio en la energía interna.
Otro ejemplo importante está relacionado con lo que hemos utilizado antes para calcular el trabajo:
w = – ∫ViVf PdV
Vemos aquí que el trabajo depende entonces de dos variables de estado. Sin embargo, todavía podemos acabar con diferentes valores para el trabajo dependiendo de cómo cambie exactamente la presión. El trabajo no depende simplemente de las presiones y volúmenes iniciales y finales.
Por último, como puedes observar en este artículo, la entropía, una variable de estado, depende del calor:
ΔS = qrev / T
Sin embargo, la entropía depende específicamente del calor absorbido o emitido en el camino reversible de un cambio de estado dado. De este modo, la entropía depende del calor de un camino específico, lo que convierte al calor reversible en una función cuasi-estatal.
Problema 1
La energía de activación es la cantidad de energía necesaria para formar un estado de transición para iniciar una reacción. El uso de catalizadores permite la formación de estados de transición alternativos con diferentes energías de activación sin cambiar el producto formado por la reacción. A partir de esta información, ¿la energía de activación es, por tanto, una variable de trayectoria o una variable de estado?
Problema 2
Puedes observar que los cristales de diamante de carbono puro tienen una longitud determinada. ¿Es la longitud una variable de estado intensiva o extensiva?
1: La energía de activación es una variable de trayectoria
2: La longitud es una variable de estado extensiva
The post Variables de estado frente a variables de trayectoria appeared first on ChemTalk.

En este tutorial de química, se introducirá el tema del calor específico. Además, aprenderás la fórmula que acompaña a este concepto y realizarás un ejemplo para resolver los cálculos matemáticos. También aprenderás una lista de la capacidad calorífica específica de varias sustancias.
¿Es fácil calentar una sustancia? ¿Necesitan todas las sustancias la misma cantidad de calor para elevar su temperatura?
Una observación del científico Joseph Black afirmaba que, para calentar masas iguales de sustancias diferentes a través del mismo intervalo de temperatura, se necesitan cantidades distintas de energía. Por cierto, Joseph Black es más conocido por sus experimentos con dióxido de carbono, gas al que llamó «aire fijo».
Esta propiedad química, conocida como calor específico, se define como la cantidad de energía térmica necesaria para elevar la temperatura de un objeto. Cuando se habla de sustancias puras en cantidades variables, se utiliza la capacidad calorífica específica de esa sustancia, que es la cantidad de calor necesaria para elevar un grado Kelvin la temperatura de un gramo de esa sustancia. El hielo tiene una capacidad calorífica específica, pero un cubito de hielo puede tener su propio calor específico. La capacidad calorífica específica suele medirse en julios por gramo por grado Celsius (J/g ℃).
Cada sustancia tiene su propia capacidad calorífica específica, que es un valor numérico que describe esta propiedad química. Por ejemplo, el calor específico del cobre es de 0.385, un valor bastante bajo. Esto significa que un trozo de cobre se calienta con bastante facilidad.
Puedes aprender más sobre la relación entre el calor y el cambio de temperatura mediante la siguiente ecuación:
Q = mcΔT
Q= heat added (Joules)
m= mass (grams)
c= specific heat (J/ g ℃)
ΔT= change in temperature (Tfinal – T initial)
*Aunque este concepto tiene que ver con la temperatura en Celsius, no pasa nada si el valor ΔT está en una unidad de temperatura diferente. Esto se debe a que la diferencia entre las dos temperaturas será la misma independientemente de la unidad.
Esta relación sólo es válida cuando no hay cambio de fase, lo que significa que la sustancia permanece en el mismo estado de la materia de principio a fin. Las pérdidas o ganancias de calor que dan lugar a un cambio de fase, como la fusión o la congelación, tienen otras ecuaciones; ¡haz clic aquí para saber más!
P: ¿Cuál es la energía, en julios, necesaria para calentar un tubo de plomo de 100 g de peso de 25℃ a 37℃? La capacidad calorífica específica del plomo es de 0.128 J/g ℃.
R: Utiliza la ecuación e introduce los números para encontrar la respuesta.
Q = mcΔT
Q = (100g) (0.128J/g℃) (37℃ – 25℃)
Q = (100) (0.128) (12) = 153.6J
Capacidades caloríficas específicas – Ejemplos
Las unidades son julios por gramo por grado Celsius.
¿Puede el calor específico ser negativo? No que sepamos en este planeta, pero posiblemente sí en algunas estrellas o nubes de gas.
La capacidad calorífica específica del agua está asociada a la unidad denominada caloría. De hecho, una caloría se define como la cantidad de energía necesaria para elevar un gramo de agua líquida un grado centígrado. La caloría fue utilizada por primera vez por Nicolas Clement a principios del siglo XIX. Procede de la palabra latina «calor», que significa calor.
El agua tiene una gran capacidad calorífica debido a las fuertes interacciones intermoleculares. De hecho, es el líquido con mayor capacidad calorífica. Como el agua es una molécula polar, con una gran diferencia de electronegatividad entre el hidrógeno y el oxígeno, se forman enlaces de hidrógeno entre los átomos de hidrógeno positivos de una molécula y los átomos de oxígeno negativos de las moléculas cercanas. Se necesita mucha energía para aflojar y luego romper los enlaces de hidrógeno entre las moléculas de agua.
Problema 1
Una muestra de 50 g de metal se calienta con 800 julios, aumentando su temperatura en 41.6℃. ¿Cuál es la identidad del metal?
Problema 2
¿Cuántos julios de energía se necesitan para elevar 500 g por 20℃?
Problema 3
Calientas una muestra de cobre de 1.2 kg a 25℃ con 900J. ¿Cuál es la temperatura final de la muestra?
1: Cobre
2: 9000J
3: 27℃
The post ¿Qué es el calor específico? appeared first on ChemTalk.
En este tutorial, aprenderás las principales características de los diagramas de fase, también llamados diagramas de cambio de fase, así como otros tipos de diagramas, y cómo interpretar y comprender su importancia.
Una transición de fase se produce cuando una sustancia cambia de un estado de la materia a otro. Existen tres estados principales de la materia: líquido, sólido y gaseoso.
Un diagrama de fases es un gráfico que ilustra las distintas fases de una sustancia en función de múltiples variables, normalmente la temperatura y la presión. El diagrama puede ayudar a demostrar cómo el cambio de estas variables afecta al estado de la materia de una sustancia concreta.
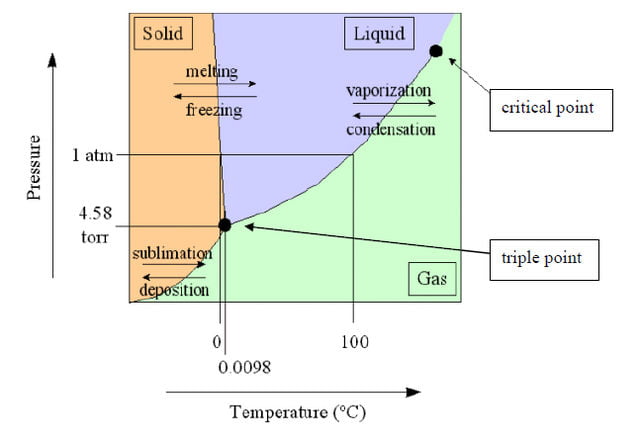
El diagrama de fases presión-temperatura es el tipo más común y básico.

Un diagrama de fases como el mostrado anteriormente representa la presión (en atmósferas) frente a la temperatura (en grados Celsius o Kelvin). Las líneas continuas representan los límites de las fases, también llamadas «curvas de coexistencia», porque hay dos fases que coexisten en equilibrio. La línea azul muestra el punto de ebullición, o transición entre estado líquido y gaseoso, la línea verde muestra el punto de congelación, o transición entre estado líquido y sólido, y la línea roja muestra las condiciones en las que un estado sólido puede convertirse directamente en fase gaseosa y viceversa.
Éstas son algunas palabras y frases comunes que oirás cuando aprendas sobre los diagramas de cambio de fase.
Curva de fusión (derretimiento o congelación): curva que muestra la transición entre los estados líquido y sólido.
Curva de vaporización (o condensación): curva que muestra la transición entre los estados gaseoso y líquido.
Curva de sublimación (o deposición): curva que muestra la transición entre los estados gaseoso y sólido.
Punto triple: punto de temperatura y presión en el que el gas, el líquido y el sólido coexisten en equilibrio.
Punto crítico: punto de temperatura y presión en el que la sustancia no distingue entre los estados líquido y gaseoso.
Límite de fase: línea que indica las condiciones en las que coexisten dos estados de la materia en equilibrio.
Un caso especial que se discute habitualmente es el diagrama de fases del agua. En el diagrama de arriba, la línea verde de puntos representa la curva de coexistencia sólido-líquido en el diagrama de fases del agua.
Podemos ver que para la sustancia media, el aumento de la presión del líquido hará que cruce la curva y se convierta en sólido. Sin embargo, en el caso del agua, el aumento de la presión del sólido (hielo) puede hacer que cruce la curva y se convierta en líquido. Este comportamiento es consecuencia de la baja densidad de la estructura cristalina del hielo, que contiene una gran cantidad de espacio libre debido a los enlaces de hidrógeno.
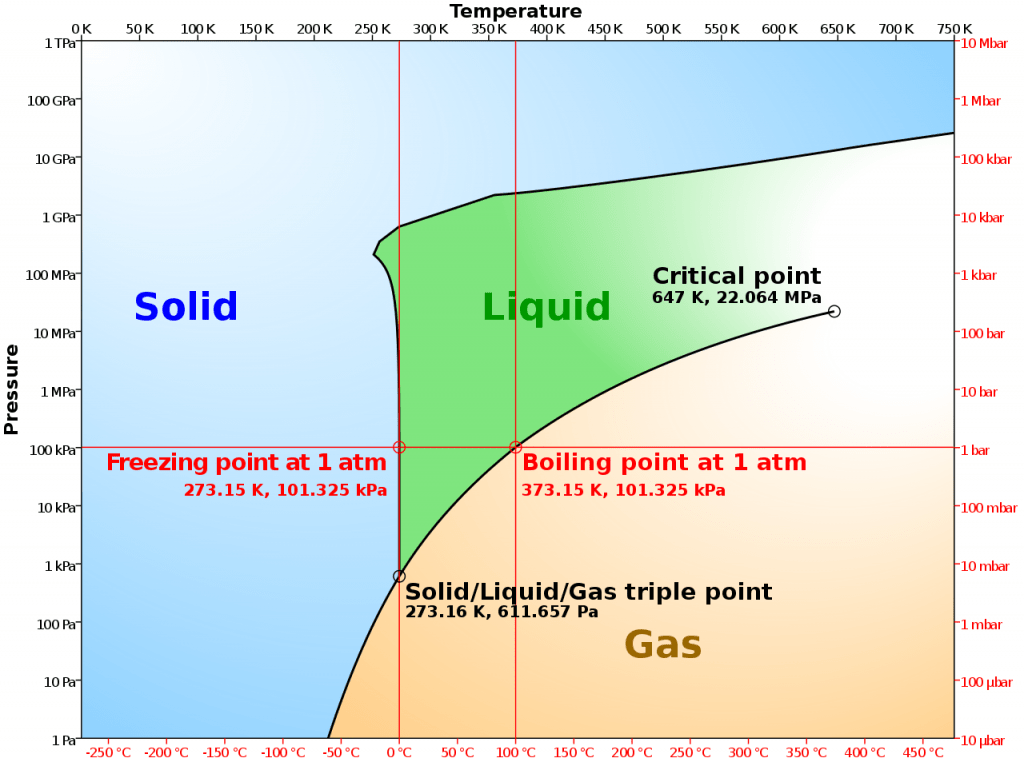
El diagrama de fases presión-temperatura no es el único. Hay muchas variables termodinámicas que pueden influir en el comportamiento de fase de una sustancia, como el volumen, la entropía, la entalpía y el potencial químico. Puedes utilizar cualquiera de estas variables para generar un diagrama de fase que muestre para qué valores de dos variables una muestra es sólida, líquida o gaseosa.
Como un diagrama bidimensional sólo puede mostrar dos variables como ejes, a veces se mostrará una tercera variable mediante curvas que muestran dónde esta variable es constante. Reciben distintos nombres según la variable que representen. Por ejemplo, una isobara muestra una presión constante, una isocorriente un volumen constante y una isoterma una temperatura constante.
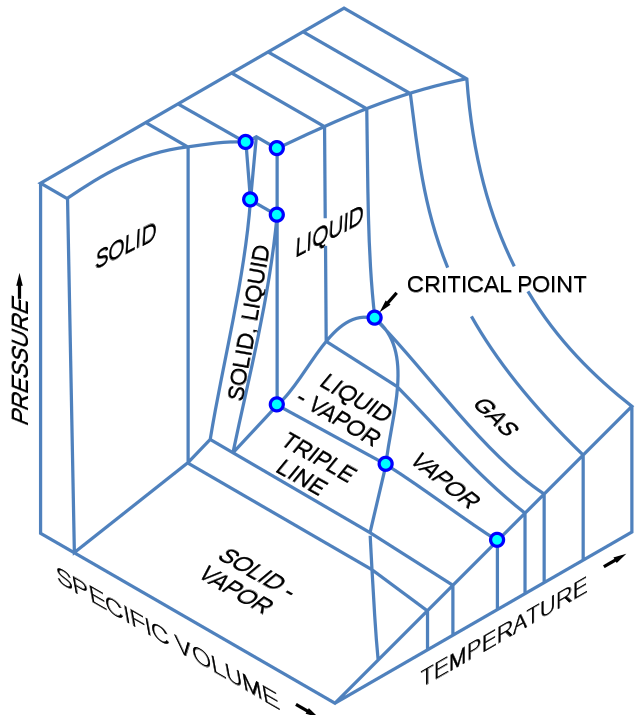
Los diagramas tridimensionales de cambio de fase trazan tres variables termodinámicas y muestran regiones del espacio correspondientes a diferentes fases. En este tipo de diagrama, tenemos una línea triple en lugar de un punto triple, y superficies de coexistencia en lugar de curvas de coexistencia. A continuación se muestra un diagrama tridimensional genérico que representa la temperatura, la presión y el volumen específico.
Las mezclas binarias tienen diagramas de fases trazados en función de la composición, normalmente representada como fracción molar de un componente. Esto muestra cómo el comportamiento de fase de la mezcla depende de la composición específica de la muestra. Una de las características más importantes de este tipo de diagrama es el punto eutéctico, que es el punto del diagrama en el que el punto de fusión es más bajo, y suele situarse en algún punto intermedio. En el diagrama siguiente, el punto eutéctico se encuentra en torno al 40 mol % de nitrato de amonio.
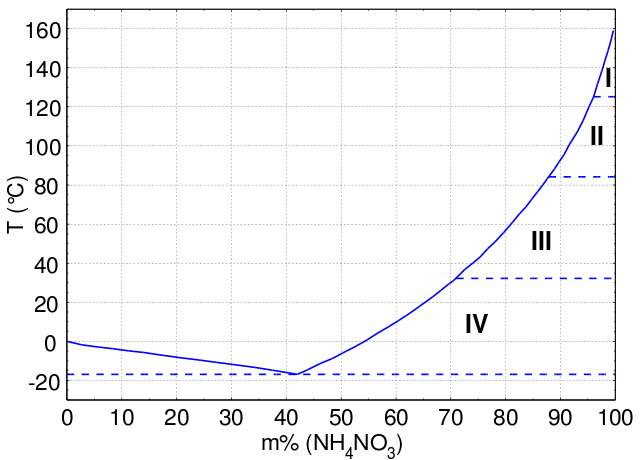
Diagrama de fases de una mezcla binaria de nitrato de amonio y agua. Por encima de la curva azul está el estado líquido, mientras que por debajo está el estado sólido.
Los sólidos cristalinos pueden tener múltiples estructuras posibles, que dependen de variables termodinámicas. Por eso puede que oigas a alguien hablar de diferentes formas de hielo, como hielo II, hielo III o hielo IV. También puede haber varios minerales con la misma fórmula química, como la calcita y el aragonito, que también son fases cristalinas diferentes (polimorfos) de la misma sustancia. En casos como éste, es habitual ver un diagrama de fases con muchas fases, todas ellas sólidas.
Un truco científico: congelar el agua al instante (en el blog de Susan Koch)
The post Diagramas de fases appeared first on ChemTalk.